集群分析
集群分析(Cluster analysis)和類別分析(Classification analysis)有很相近的關係,但用法不同。一般來說,如果你有已經有一筆資料並清楚其類別,想要對新的資料但不清楚其類別時,可以做類別分析。但如果你只有一筆資料,但並不清楚其原有的分類,或是根本就沒有分類那時我們就會考慮用集群分析。透過每個樣本點與其它樣本點所定義出來的距離不斷的聚合(merge)資料,最後聚成一串資料,如圖一所示。(長得和分類樹很像但是兩者是完全不同的東西。)
主要的想法就是對每個樣本點和其它的樣本點定義距離[1],根據距離的大小來決定聚合的順序。
下面是一個五個點的例子:合併後的子群間的距離用 complete (最長的距離)來計算。
1. 根據樣本點的距離矩陣找到最小的距離,再把最小距離的兩個點合併。例如這個最小的距離是0.5所以我們把A、E合併。
|
|
|
A |
B |
C |
D |
|
|
B |
|
1 |
|
|
|
|
|
C |
|
1.5 |
0.6 |
|
|
|
|
D |
|
2 |
1.7 |
1.1 |
|
|
|
E |
|
0.5 |
0.8 |
1.9 |
0.7 |
|
2. 合併矩陣時,(A,E)對其它各點的新距離是取 A 或 E 對其它各點距離的最大值(complete 法)。例如 (A,E) 和 B 的距離就是
max( |A-B| , |B-E| ) = max( 1 , 0.8 ) = 1。
|
|
|
A,E |
B |
C |
|
|
B |
|
1 |
|
|
|
|
C |
|
1.9 |
0.6 |
|
|
|
D |
|
2 |
1.7 |
1.1 |
|
3. 同樣的再根據新的矩陣來找最小值,這次找到的是0.6。新的合併矩陣如下。
|
|
|
A,E |
B,C |
|
|
B,C |
|
1.9 |
|
|
|
D |
|
2 |
1.7 |
|
4. 用同樣的方法就可以把整個樣本點合併完。
|
|
|
A,E |
|
|
(B,C),D |
|
2 |
|
最後的合併結果就是:((B,C),D),(A,E))
我們可以把距離矩陣丟去R裡面去執行。
>m #原來的距離矩陣
|
|
[,1] |
[,2] |
[,3] |
[,4] |
[,5] |
|
[1,] |
0 |
1 |
1.5 |
2 |
0.5 |
|
[2,] |
1 |
0 |
0.6 |
1.7 |
0.8 |
|
[3,] |
1.5 |
0.6 |
0 |
1.1 |
1.9 |
|
[4,] |
2 |
1.7 |
1.1 |
0 |
0.7 |
|
[5,] |
0.5 |
0.8 |
1.9 |
0.7 |
0 |
> rownames(m)=LETTERS[1:5]
> colnames(m)=LETTERS[1:5] #加上各行列的名字
> m
|
|
A |
B |
C |
D |
E |
|
A |
0 |
1 |
1.5 |
2 |
0.5 |
|
B |
1 |
0 |
0.6 |
1.7 |
0.8 |
|
C |
1.5 |
0.6 |
0 |
1.1 |
1.9 |
|
D |
2 |
1.7 |
1.1 |
0 |
0.7 |
|
E |
0.5 |
0.8 |
1.9 |
0.7 |
0 |
>md=as.dist(m,diag=F,upper=F);md #定成dist(距離)的物件
|
|
A |
B |
C |
D |
|
B |
1 |
|
|
|
|
C |
1.5 |
0.6 |
|
|
|
D |
2 |
1.7 |
1.1 |
|
|
E |
0.5 |
0.8 |
1.9 |
0.7 |
> cm=hclust(md) #用cluster做集群
> plot(cm,xlab="made data",sub="Merge method:Complete",axes=F);box();
> axis(2,at=c(0.5,0.6,1.7,2),cex.axis=0.8)
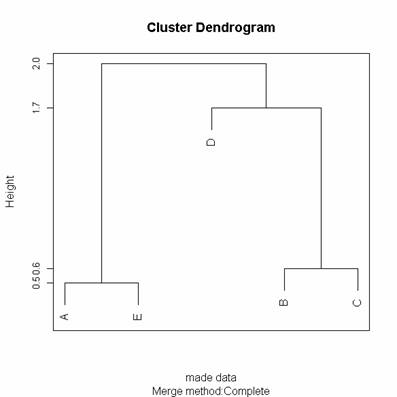
圖一、用cluster做集群後的結果,和之前的推算一樣。
現在來對比較複雜的iris data來做集群。
>data(iris)
?hclust #可以查一下我們會用到的指令
?dist
>diris=dist(iris[,-5])
>ciris=hclust(diris);ciris
> ciris
Call:
hclust(d = diris)
Cluster method : complete
Distance : euclidean
Number of objects: 150
> names(ciris)
[1] "merge" "height" "order" "labels" "method"
[6] "call" "dist.method"
>ciris$merge
|
#有減號的代表是樣本點,沒有的代表是聚合後的子群[ ](圖二中紅字的部分)。例如子群[1,]是樣本點102和143聚合而成。[133,]是子群114和125聚合而成。[138,]是子群[133,]和樣本點42聚合而成。如圖二所示。 |
>plot(ciris,cex=0.5,xlab="Iris",sub="Merge method:Complete");box()
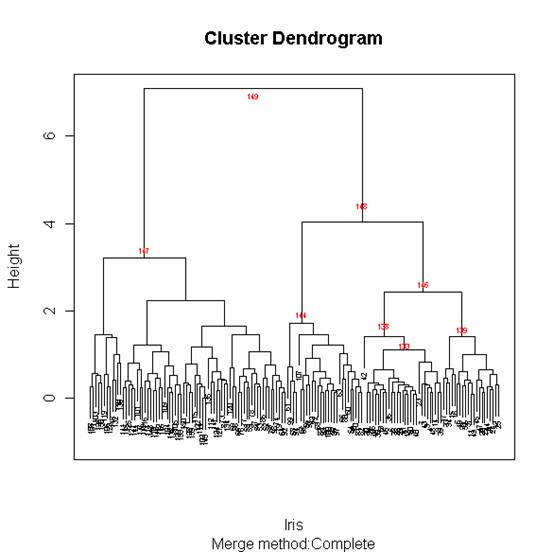
圖二、 用complete法來聚合Iris data
如果我們想把資料分群的話:
>memb = cutree(ciris, k = 3) #分三群
>fmemb=factor(memb,labels=c("setosa","virginica","versicolor"))
> table(fmemb,iris[,5])
|
|
|
Iris[,5] |
||
|
|
|
setosa |
versicolor |
virginica |
|
fmemb |
setosa |
50 |
0 |
0 |
|
virginica |
0 |
23 |
49 |
|
|
versicolor |
0 |
27 |
1 |
|
看起來是沒有分得比LDA 或是CART來得好,因為LDA 或是CART是找到可以分得最開的變數的線性組合,或是根據每一個變數去看,而集群只有把所以的變數都弄成一個量來看,所以有些距離一樣(可能各分量不同)的樣本點就被疊在一塊以致於很難分開。除非我們可以定義更有效的距離矩陣。
集群分析除了可以聚合個體(individual 或是subject)之外也可以聚合變數。只要定義好距離就可以。在基因資料中,由於變數相當多而受試者相對稀少,一般的統計方法幾乎沒有辦法使用。這時候集群分析就可以派上用場。雖然我們也不知道在每群中那些變數是重要的,但是可以做一些初步的歸納,再做進一步的實驗。